More about annotations¶
When running prepare-rpbp-genome, Rp-Bp extracts all putative Ribo-seq ORFs and assigns to each one a label based on its position relative to the annotated transcript-exon structure. Labels can be useful to quickly identify certain ORF types that may be of particular interest, e.g. upstream ORFs, or ORFs from non-coding RNAs.
Note
The ORF “id” is of the form transcript_seqname:start-end:strand. seqname is the chromosome or contig. The start codon is included, but the stop codon is not. The host transcript “id” should not contain underscores!
Hint
In some cases, the ORF label may not be consistent with the host transcript, as reported by the ORF “id”. To resolve such seemingly incoherent assignments, compatible transcripts are reported for each ORF in <genome_name>.orfs-labels.annotated[.orf_note].tab.gz and shown in the prediction dashboard (see Visualization and QC).
Categories of Ribo-seq ORFs¶
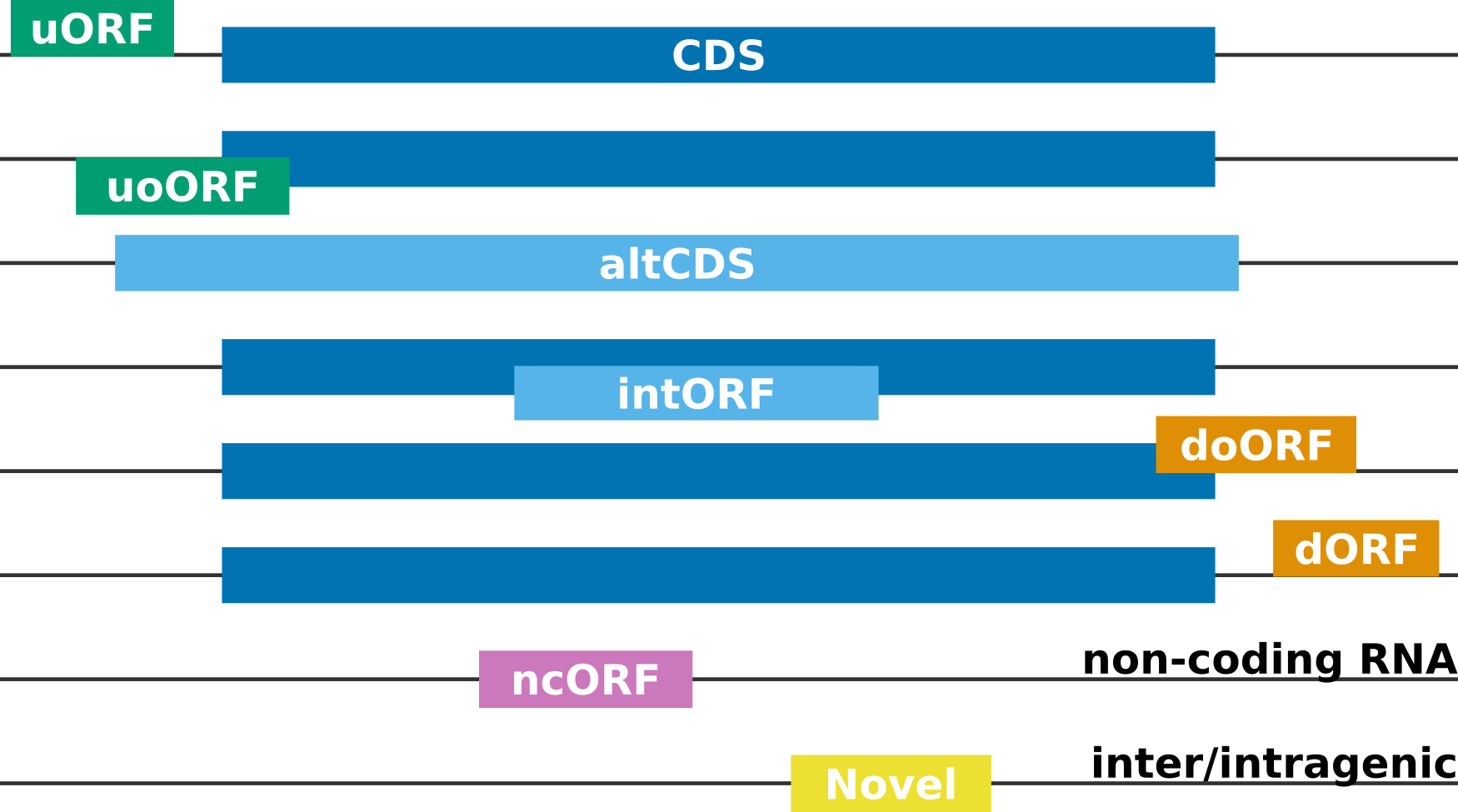
CDS: Canonical (annotated) coding sequence
altCDS: Alternative CDS (e.g. N/C-terminus extension/truncation, alternatively spliced variants, etc.)
intORF: Translation event within a CDS (in- or out-of-frame)
uORF/uoORF: Translation event in the 5’ untranslated region (UTR) of or partially overlapping an annotated protein-coding gene
dORF/doORF: Translation event in the 3’ untranslated region (UTR) of or partially overlapping an annotated protein-coding gene
ncORF: Translation event in an RNA annotated as non-coding (lncRNA, pseudogene, etc.)
Novel: Translation event inter- or intragenic (only when Rp-Bp is run with a de novo assembly, see below)
Labels such as overlap or suspect arise when Rp-Bp is not able to resolve the position of an ORF without ambiguity. In practice, we do not see these categories for standard organisms.
More about de novo ORF discovery¶
It is often of interest to identify Novel Ribo-seq ORFs, i.e. not only un-annotated ORFs, but ORFs that do not overlap the annotation at all. For Rp-Bp, there is no difference between annotated and de novo assembled transcripts. In both cases, ORFs are extracted from the transcripts based on the given start and stop codons. Hence, when matching RNA-seq is available, we highly recommend to create a de novo assembly. The only requirement is that the assembler produces a valid GTF file (or a format that can be converted to GTF).
Caution
In a de novo assembly, coding regions are typically not identified (that is what Ribo-seq is for!). However, if your assembly also includes CDS annotations, they must satisfy the start/stop codon GFF2 specifications (stop codon not included in the CDS.)
Hint
prepare-rpbp-genome may fail unexpectedly with ERROR: Duplicate ORF ids were found. Duplicate ORF ids can arise due to different ORFs having the same start/end coordinates and the same host transcript, or when a de novo assembled transcript has a complete, exact match of the intron chain with an existing, annotated transcript. This usually only affects a handful of transcripts. To handle this situation, you can identify duplicate ORF ids in <genome_name>.orfs-genomic[.orf_note].annotated.bed.gz and <genome_name>.orfs-genomic[.orf_note].de-novo.bed.gz, collect the associated host transcript ids into a file, remove them from the newly created de novo GTF .e.g with grep -v -f duplicated_transcripts.txt path/to/de-novo.gtf > path/to/de-novo.slim.gtf, update de_novo_gtf in the configuration file, and re-run prepare-rpbp-genome.
Tip
When matching RNA-seq is available, you can also perform translational efficiency analysis (TEA), see Ribotools for more information.